Ingesting mouse scRNA-Seq data with Panpipes
Let’s run through an example of reading a mouse scRNA-Seq dataset into MuData and computing QC metrics using Panpipes. The workflow describes the steps run by the pipeline in greater detail.
For all the tutorials, we will append the --local command which ensures that the pipeline runs on the computing node you’re currently on, namely your local machine or an interactive session on a computing node on a cluster.
Directories and data
Let’s create a main ingest directory and in that directory, the directory data.
mkdir ingest ingest/data
In this tutorial, we will use a mouse lymph node 10X dataset. To download the dataset into the data folder, you can use the following commands:
cd ingest/data
curl https://cf.10xgenomics.com/samples/cell-exp/7.2.0/4plex_mouse_LymphNode_Spleen_TotalSeqC_multiplex_LymphNode1_BC1_AB1/4plex_mouse_LymphNode_Spleen_TotalSeqC_multiplex_LymphNode1_BC1_AB1_count_sample_filtered_feature_bc_matrix.h5 -o filtered_feature_bc_matrix.h5
The ingest directory should now have the following structure:
ingest
└── data
└── filtered_feature_bc_matrix.h5
Edit submission and yaml file
Create a submission file like the one we provide. The submission file specifies the type of input data and its location in the file system. For more information about the submission file generation please refer to the usage guidelines.
The ingest workflow provides the possibility to compute percentages and scores (sc.tl.score_genes) of user-specified gene groups and to plot them in violin plots. For that, a gene list csv-file needs to be provided by the user. The usage guidelines describe the format of the gene list in detail. For this tutorial, you can use the provided gene list that contains mouse genes of the following groups: hb, exclude, ig, rp, mt, CD8cytotoxic.
After creating the submission and gene list files, call panpipes ingest config in the ingest directory (you potentially need to activate the conda environment with conda activate pipeline_env first!). This will generate a pipeline.log and a pipeline.yml file.
Modify the pipeline.yml or simply replace it with the provided yaml. Make sure to specify the correct path to the submission file. If you’re using the provided example yaml file, you potentially need to add the path of the conda environment in the yaml.
Run Panpipes
In ingest, run panpipes ingest make full --local to ingest your dataset.
In case you used the provided yaml, you should now have the following files in ingest:
ingestion
├── data
│ └── filtered_feature_bc_matrix.h5
├── figures
│ └── rna
│ ├── scatter_sample_id_rna-log1p_nUMI_v_rna-log1p_genes.png
│ ├── scatter_sample_id_rna-nUMI_v_rna-genes.png
│ ├── scatter_sample_id_rna-nUMI_v_rna-pct_mt.png
│ ├── violin_sample_id_rna-n_genes_by_counts.png
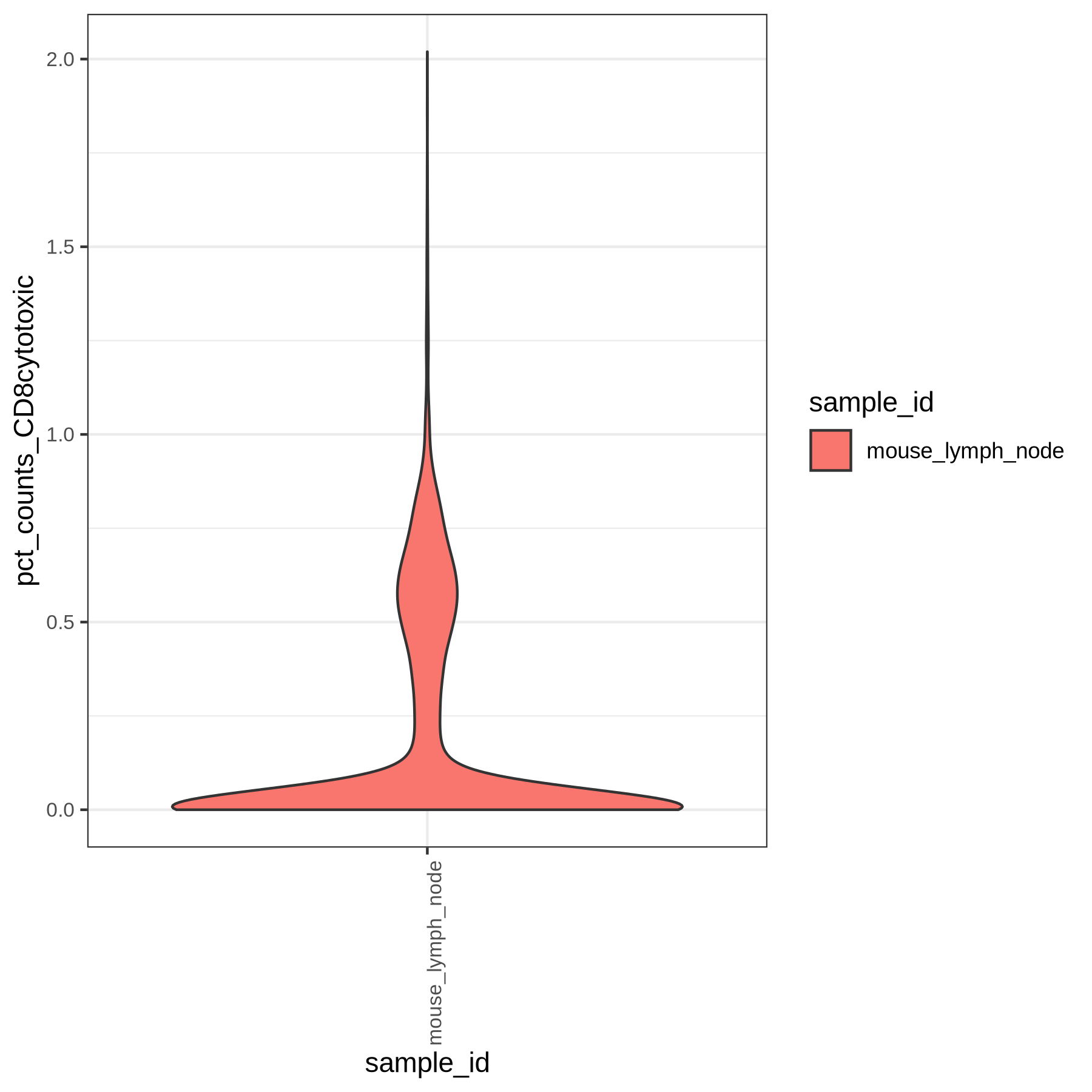
│ ├── violin_sample_id_rna-pct_counts_CD8cytotoxic.png
│ ├── violin_sample_id_rna-pct_counts_hb.png
│ ├── violin_sample_id_rna-pct_counts_mt.png
│ ├── violin_sample_id_rna-pct_counts_rp.png
│ └── violin_sample_id_rna-total_counts.png
├── logs
│ ├── concat_filtered_mudatas.log
│ ├── plot_qc.log
│ ├── load_mudatas_mouse_lymph_node.log
│ └── run_scanpy_qc_rna.log
├── tmp #MuData without QC metrics
│ └── mouse_lymph_node.h5mu
├── pipeline.log
├── pipeline.yml
├── sample_submission_file.txt
├── mouse_lymph_node_cell_metadata.tsv # Metadata, i.e. .obs
└── mouse_lymph_node_unfilt.h5mu
The final MuData object with computed QC metrics is ingest/mouse_lymph_node_unfilt.h5mu. A MuData object without QC metrics is also available and stored in the tmp folder. The metadata of the final Mudata object is additionally extracted and saved as a tsv file, mouse_lymph_node_cell_metadata.tsv.
With the plots in ingest/figures/rna you can now decide on cutoffs for filtering. The plots include scatter as well as violin plots of the QC metrics specified in the pipeline.yml.

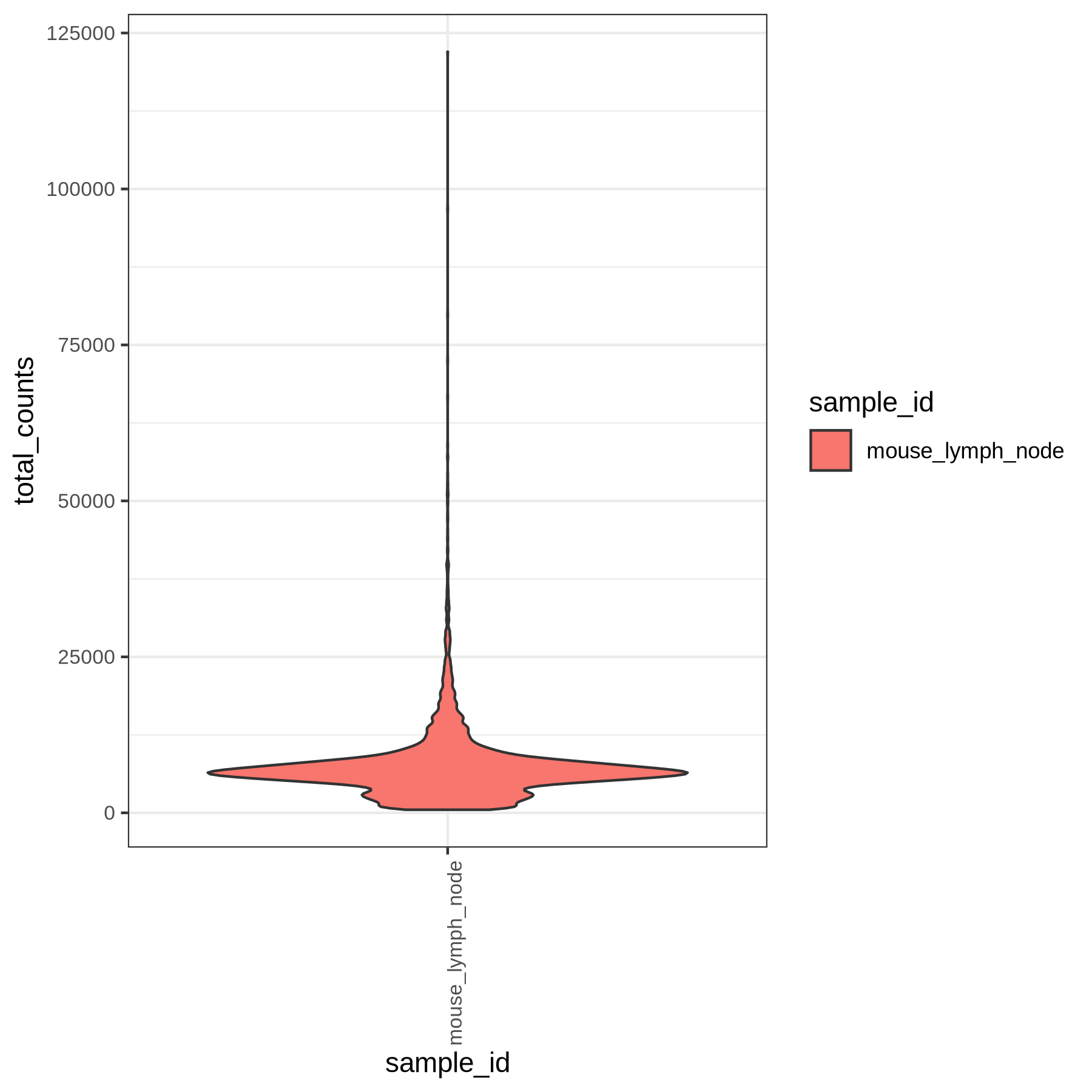
Violin plots of the computed gene group percentages and gene group scores are generated by the pipeline (if specified in the yaml):

We have demonstrated a use case of ingesting mouse dataset leveraging a custom gene file generated to allow panpipes’ actions to be performed. Users can play with the actions and follow the standard analysis steps as demonstrated for human data in panpipes preprocess(../filtering_data/filtering_data_with_panpipes.md)
Note: We find that keeping the suggested directory structure (one main directory by project with all the individual steps in separate folders) is useful for project management. You can of course customize your directories as you prefer, and change the paths accordingly in the pipeline.yml config files!