Reference mapping
scvi-tools has implemented some changes and as a result old models (scvi-tools<=1.0) require pandas <2.0.
https://github.com/scverse/scvi-tools/issues/2387
This will be fixed in a future version of scvi-tools.
To use an old model in panpipes refmap please downgrade pandas.
Reference mapping is the process of aligning a new single cell dataset to an existing single cell atlas generated with a deep learning model that allows to see the new data (referred to as query) in the context of a pre-genererated latent space (normally called reference).
Panpipes supports the use of scvi-tools for constructing and leveraging reference models to do query to reference mapping (scvi, scanvi and TotalVI).
We will show how panpipes allows to map the same query to two different references models, an scvi and a scanvi reference model, respectively. In this case, the underlying data used for the two reference models is exactly the same, but we’re going to use it just as an example of panpipes’ functionality to map the same query in parallel to two reference models.
Download the data for the reference and query used in this example.
Now create a directory where you will run the refmap workflow:
mkdir reference_mapping && cd reference_mapping
mkdir data.dir
mkdir models
ensure the data and the models you downloaded are in the data.dir and models folders respectively
Please note that the we shared the model files with two separate models, but scvi-tools require them to be supplied as model.pt instead of a custom name. This is not a requirement of panpipes, but a standard requirement when dealing with scvi-tools pytorch models. We therefore create two folders, one for the scvi and one for the scanvi models and save the two models in each folder. The name you use for each of them will be reused by the pipeline to save model-specific outputs, so make them as informative as you can!
ls data.dir
pancreas_querydata.h5ad
pancreas_refdata.h5mu
ls models
pancreas_model/model.pt
pancreas_model_scanvi/model.pt
As with all the other worflows you can configure the refmap workflow using:
panpipes refmap config
which will generate the pipeline.yml file that is used as input.
We provide the pipeline yml file for this tutorial here.
In the config file, we only have to specify the path to the query, if the query has a batch covariate and if it has a celltype annotation column:
#----------------------
# query dataset
#----------------------
query: pancreas_query.h5ad
# we support raw10x formats, or preprocessed quality filtered mudata or anndata as input query
modality: rna
# if you supplied a mu data, specify the modality to be used
# only RNA modality supported by current models!
# does your query data have a batch effect, what column?
query_batch:
# does your query have a celltype annotation you want to use to compare to
# the transferred label?
# leave empty if not
query_celltype: celltype
For the reference models, we specify the path to each model, and since we have the reference data we can also supply it.
#----------------------
# scvi tools params
#----------------------
# specify one or more reference models that you would like to use as reference
# you can use your own reference that you built using pipeline_integration here
# leave blank for no model specification
# specify the reference anndata/mudata with RNA. you need this only if you want to calc umap on both reference and query data.
# otherwise leave blank and only provide model paths
reference_data: path_to_mudata
[...]
# Specify the full path to the model, i.e. Path/to/scanvi/model.pt
# if not running the method, remove the dummy - path_to_xxx and leave blank
scvi:
- path_to_scvi
scanvi:
- path_to_scanvi
Panpipes doesn’t require the reference anndata/mudata to run, the reference models are sufficient. This may be a common scenario for many users, for example when the reference is too big to be shared or there are some privacy issues and the data can’t be shared.
However, if the reference data is supplied, panpipes refmap will use it to update the latent representation and plot the query and reference cells in the same umap.
You can run the workflow with
panpipes refmap make full
For the dataset we’re using it should not take longer than 20 minutes to generate the outputs. Let’s take a look:
tree reference_mapping
├── data.dir
│ ├── pancreas_querydata.h5ad
│ └── pancreas_refdata.h5mu
├── figures
│ ├── SCANVI_predicted_vs_observed_labels_query_data.png
│ ├── umap_pancreasmodel_X_scVI.png
│ └── umap_pancreasmodelscanvi_X_scANVI.png
├── logs
│ ├── pancreas_model.log
│ ├── pancreas_model_scanvi.log
│ ├── refmapscib_pancreasmodel_X_scvi.log
│ ├── refmapscib_pancreasmodelscanvi_X_scanvi.log
│ └── setup_dirs.sentinel
├── models
│ ├── pancreas_model
│ └── pancreas_model_scanvi
├── pipeline.log
├── pipeline.yml
└── refmap
├── query_to_reference_pancreasmodel_X_scvi.h5mu
├── query_to_reference_pancreasmodelscanvi_X_scanvi.h5mu
├── scib.query_pancreasmodel_X_scvi.csv
├── scib.query_pancreasmodelscanvi_X_scanvi.csv
├── umap_pancreasmodel_X_scvi.csv
└── umap_pancreasmodelscanvi_X_scanvi.csv
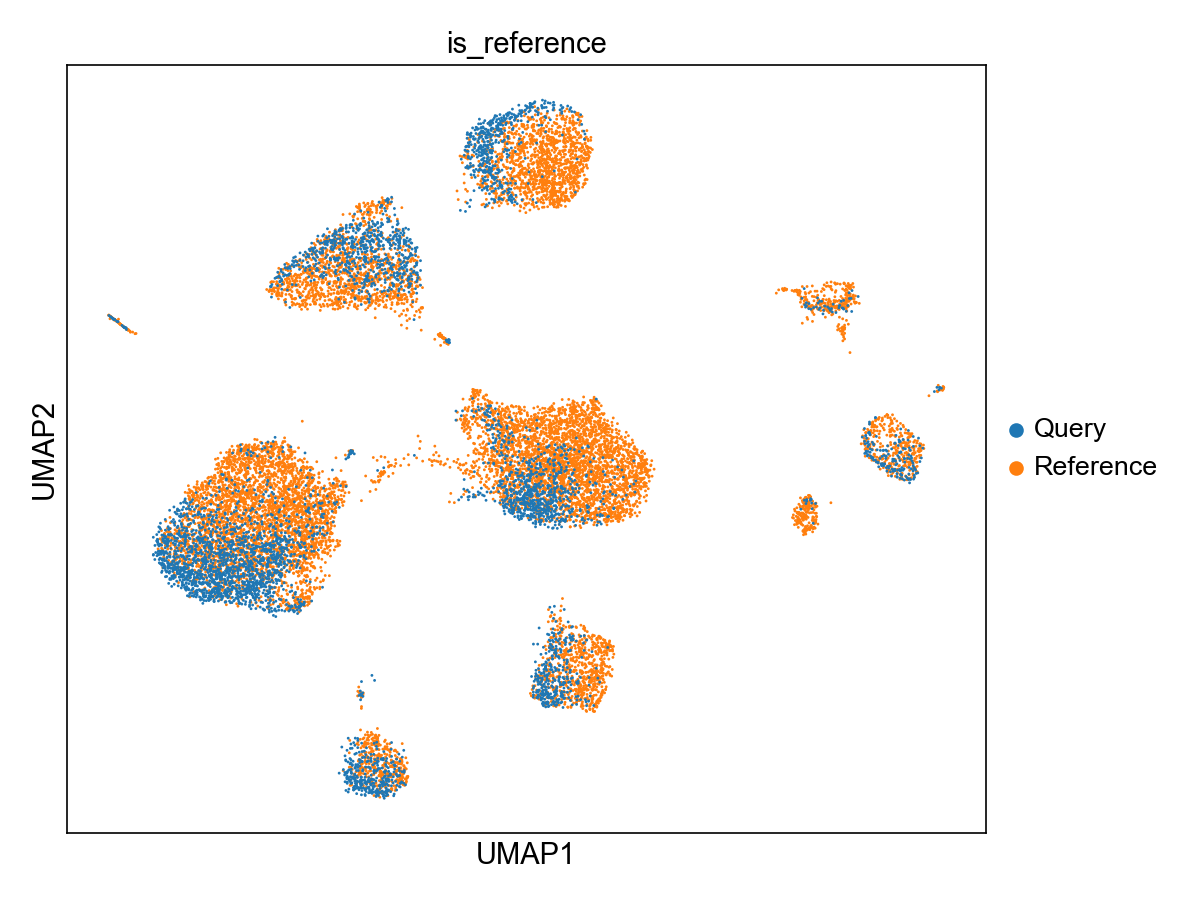
In the figures folder, you will see that panpipes has produced the umaps for both scvi and scanvi query to reference mapping runs:
The scvi Umap showing query and reference in the same representation

The scanVI Umap showing query and reference in the same representation
For the scanvi run, which transfers the labels present in the reference to the query, we can plot a confusion matrix which compares the observed query labels with the predicted labels.

Besides figures, refmap also outputs other useful files, in the refmap directory:
file |
file type |
info |
|---|---|---|
query_to_reference_pancreasmodel_X_scvi.h5mu |
mudata |
the concatenated query and reference data for the scvi run |
query_to_reference_pancreasmodelscanvi_X_scanvi.h5mu |
mudata |
the concatenated query and reference data for the scanvi run |
scib.query_pancreasmodel_X_scvi.csv |
csv |
scib metrics results for the scvi run |
scib.query_pancreasmodelscanvi_X_scanvi.csv |
csv |
scib metrics results for the scvi run |
umap_pancreasmodel_X_scvi.csv |
csv |
umap coordinates |
umap_pancreasmodelscanvi_X_scanvi.csv |
csv |
umap coordinates |
Please note that panpipes supports the use of both h5ad or h5mu input data for query and reference, but we save the outputs as h5mu data.
Useful notes
The reference mapping models require that the query is formatted to match the features in the reference. It is therefore common practice to share the list of the highly variable genes (or the equivalent set of features) that was used to build the reference, alongside the model itself. We had already formatted the data for this tutorial, but if you’re interested in using your own query you should pay special attention to this.
Checkout the workflows section of the documentation for more info on how to format your query data.